Description: Homo sapiens Wiskott-Aldrich syndrome (WAS), mRNA. RefSeq Summary (NM_000377): The Wiskott-Aldrich syndrome (WAS) family of proteins share similar domain structure, and are involved in transduction of signals from receptors on the cell surface to the actin cytoskeleton. The presence of a number of different motifs suggests that they are regulated by a number of different stimuli, and interact with multiple proteins. Recent studies have demonstrated that these proteins, directly or indirectly, associate with the small GTPase, Cdc42, known to regulate formation of actin filaments, and the cytoskeletal organizing complex, Arp2/3. Wiskott-Aldrich syndrome is a rare, inherited, X-linked, recessive disease characterized by immune dysregulation and microthrombocytopenia, and is caused by mutations in the WAS gene. The WAS gene product is a cytoplasmic protein, expressed exclusively in hematopoietic cells, which show signalling and cytoskeletal abnormalities in WAS patients. A transcript variant arising as a result of alternative promoter usage, and containing a different 5' UTR sequence, has been described, however, its full-length nature is not known. [provided by RefSeq, Jul 2008]. Transcript (Including UTRs) Position: hg19 chrX:48,542,186-48,549,817 Size: 7,632 Total Exon Count: 12 Strand: + Coding Region Position: hg19 chrX:48,542,243-48,549,553 Size: 7,311 Coding Exon Count: 12
ID:WASP_HUMAN DESCRIPTION: RecName: Full=Wiskott-Aldrich syndrome protein; Short=WASp; FUNCTION: Effector protein for Rho-type GTPases. Regulates actin filament reorganization via its interaction with the Arp2/3 complex. Important for efficient actin polymerization. Possible regulator of lymphocyte and platelet function. Mediates actin filament reorganization and the formation of actin pedestals upon infection by pathogenic bacteria. SUBUNIT: Interacts with NCK1 (via SH3 domains) (By similarity). Interacts with CDC42, RAC, NCK, HCK, FYN, SRC kinase FGR, BTK, ABL1, PSTPIP1, WIP, and to the p85 subunit of PLC-gamma. Binds the Arp2/3 complex. Interacts (via C-terminus) with ALDOA. Interacts with E.coli effector protein EspF(U). INTERACTION: Self; NbExp=3; IntAct=EBI-346375, EBI-346375; P60953:CDC42; NbExp=10; IntAct=EBI-346375, EBI-81752; Q14247:CTTN; NbExp=3; IntAct=EBI-346375, EBI-351886; P08631:HCK; NbExp=9; IntAct=EBI-346375, EBI-346340; O43516:WIPF1; NbExp=11; IntAct=EBI-346375, EBI-346356; SUBCELLULAR LOCATION: Cytoplasm, cytoskeleton. TISSUE SPECIFICITY: Expressed predominantly in the thymus. Also found, to a much lesser extent, in the spleen. DOMAIN: The WH1 (Wasp homology 1) domain may bind a Pro-rich ligand. DOMAIN: The CRIB (Cdc42/Rac-interactive-binding) region binds to the C-terminal WH2 domain in the autoinhibited state of the protein. Binding of Rho-type GTPases to the CRIB induces a conformation change and leads to activation. PTM: Phosphorylated at Tyr-291 by FYN and HCK, inducing WAS effector activity after TCR engagement. Phosphorylation at Tyr-291 enhances WAS activity in promoting actin polymerization and filopodia formation. DISEASE: Defects in WAS are the cause of Wiskott-Aldrich syndrome (WAS) [MIM:301000]; also known as eczema-thrombocytopenia- immunodeficiency syndrome. WAS is an X-linked recessive immunodeficiency characterized by eczema, thrombocytopenia, recurrent infections, and bloody diarrhea. Death usually occurs before age 10. DISEASE: Defects in WAS are the cause of thrombocytopenia type 1 (THC1) [MIM:313900]. Thrombocytopenia is defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. DISEASE: Defects in WAS are a cause of neutropenia severe congenital X-linked (XLN) [MIM:300299]. XLN is an immunodeficiency syndrome characterized by recurrent major bacterial infections, severe congenital neutropenia, and monocytopenia. SIMILARITY: Contains 1 CRIB domain. SIMILARITY: Contains 1 WH1 domain. SIMILARITY: Contains 1 WH2 domain. SEQUENCE CAUTION: Sequence=AAH02961.1; Type=Erroneous initiation; WEB RESOURCE: Name=WASbase; Note=WAS mutation db; URL="http://bioinf.uta.fi/WASbase/"; WEB RESOURCE: Name=WASPbase; Note=WAS mutation db; URL="http://homepage.mac.com/kohsukeimai/wasp/WASPbase.html"; WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/WAS"; WEB RESOURCE: Name=Wikipedia; Note=Wiskott-Aldrich syndrome protein entry; URL="http://en.wikipedia.org/wiki/Wiskott-Aldrich_syndrome_protein";
To design primers for a non-coding sequence, zoom to a region of interest and select from the drop-down menu: View > In External Tools > Primer3
Genetic Association Studies of Complex Diseases and Disorders
Genetic Association Database (archive): WAS CDC HuGE Published Literature: WAS Positive Disease Associations: X-linked thrombocytopenia Related Studies:
X-linked thrombocytopenia de Saint Basile G et al. 1996, Isolated X-linked thrombocytopenia in two unrelated families is associated with point mutations in the Wiskott-Aldrich syndrome protein gene., The Journal of pediatrics. 1996 Jul;129(1):56-62.
[PubMed 8757563]
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
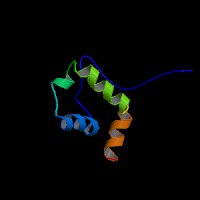
ModBase Predicted Comparative 3D Structure on P42768
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.